Tutorials,
BRB-seq
| 31 May, 2022
Bulk RNA barcoding and sequencing (BRB-seq) is an innovative, efficient, and cost-effective RNA sequencing technology that leverages the benefits method of early-stage barcoding and unique molecular identifiers (UMIs) to produce consistent, reproducible, and uniform RNA sequencing data (Alpern et al. 2019). Despite the utility of RNA sequencing (RNA-seq) in transcriptomics, traditional RNA-seq technologies are still too time consuming, costly, and analytically challenging to replace reverse transcription-quantitative real-time PCR (qRT-PCR) as the preferred method of quantifying changes in gene expression. In contrast, BRB-seq generates high-quality transcriptomic data with mere hours of hands-on time at a cost tantamount to profiling four individual genes using conventional qRT-PCR (Alpern et al. 2019).
Sample barcoding and early multiplexing first emerged in the field of single-cell transcriptomics as a way to reduce cost and handling time by generating a single sequencing library that contains multiple distinct samples (Ziegenhain et al. 2017). While other methods have attempted to address the shortcomings in bulk RNA sequencing, they tend to result in biased libraries or miss the mark on cost-effectiveness and handling time (Alpern et al. 2019). BRB-seq fills this previously unmet need by applying sample barcoding and early multiplexing principles to bulk RNA profiling, a highly optimized and rigorously evaluated process that matches the performance of far more costly methods and outperforms the best single-cell RNA-seq protocol, SCRB-seq (Alpern et al. 2019). Although BRB-seq does not allow for the analysis of full-length transcripts, splice variants, fusion genes, or address research questions involving RNA editing, it is arguably unnecessary in most cases to generate a sequencing library of complete transcripts.
The BRB-seq Innovation
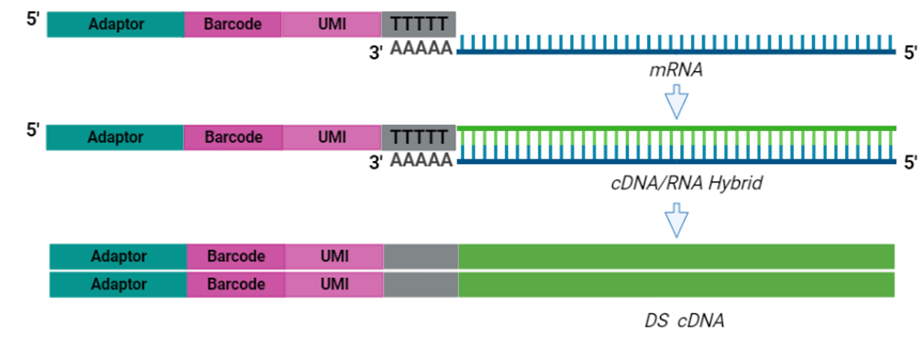
The cornerstone of BRB-seq technology is the use of highly optimized barcoded oligo(dT) primers that uniquely tag each individual RNA sample during the first-strand synthesis step of cDNA library preparation. The barcoded nucleotide sequence includes an adaptor for primer annealing, a 14-nt long barcode that assigns a unique identifier to each individual RNA sample, and a random 14-nt long UMI that tags each mRNA molecule with a unique identifier to distinguish between original mRNA transcripts and duplicates that result from amplification. Furthermore, BRB-seq allows individually barcoded RNA samples to be pooled, thereby streamlining subsequent steps in cDNA library preparation and sequencing (Figure 1), while the use of UMIs decreases amplification noise by eliminating PCR amplification bias (Alpern et al. 2019; Sena et al. 2018; Ziegenhain et al. 2017).
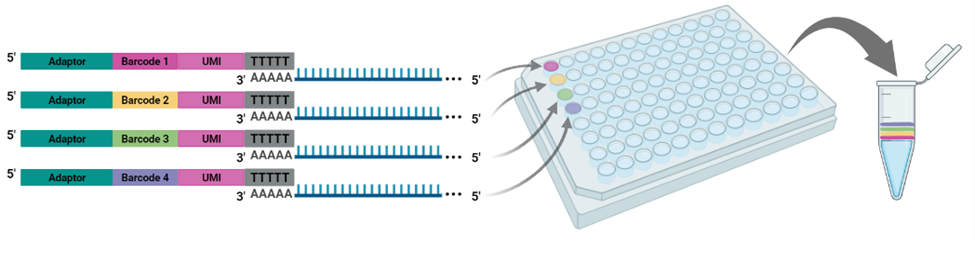
Figure 1. Each well of the BRB-seq oligo(dT) primer plate contains a distinct barcode adaptor to uniquely label the RNA sample added to each well. After reverse transcription, barcoded samples are pooled for the remainder of the cDNA library preparation workflow and library sequencing.
BRB-seq Workflow
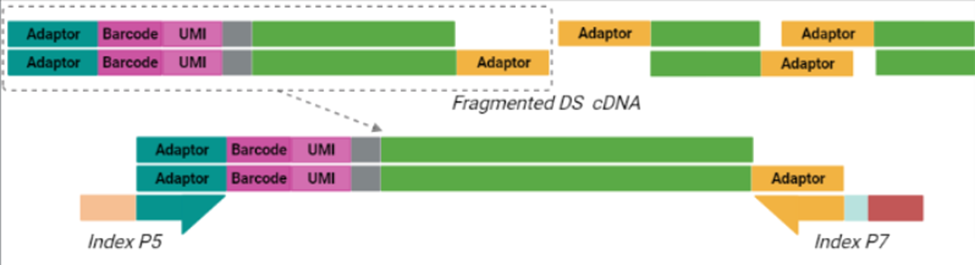
The BRB-seq protocol generates high-quality sequencing data using 10—500 ng of total purified RNA per sample. The first step—first strand synthesis via reverse transcription—generates barcoded samples with UMIs. During this step, individual barcoded samples are pooled as previously described, free primers are digested, and double-stranded full-length cDNA is generated and subsequently purified solid-phase reversible immobilization (SPRI) beads.
Purified cDNA is then subject to a process called tagmentation, in which double-stranded, full-length cDNA is fragmented and tagged using Tn5 transposase, which cleaves the cDNA and ligates adaptors for library amplification in a 7-minute reaction (Picelli et al. 2014). The resulting cDNA library is then indexed and amplified using a unique dual indexing (UDI) strategy (Index P5 and Index P7 below), which minimizes the risk of barcode misassignment after next-generation sequencing (NGS). In BRB-seq, only the tagmented fragments with both adaptors (teal and yellow below) are amplified moving forward. The 5’ terminal fragments are amplified using the Illumina-compatible UDI adaptor primers to generate UDI libraries. Different UDI primer pairs can be used to prepare and sequence multiple individual libraries in a single flow cell. After purification, the final cDNA library is ready for sequencing.
Applications of BRB-seq
BRB-seq has the potential to revolutionize the field of transcriptomics by enabling genome-wide expression analyses of numerous samples simultaneously, efficiently, and cost-effectively. Currently, up to 384 samples can be prepared by a single person in one day for less than $10/sample. This innovative approach to RNA-seq has been used in a global transcriptome analysis of brite adipocytes (Bast‐Habersbrunner et al. 2021), to evaluate the expression of mitochondrial haplotypes in Drosophila (Bevers et al. 2019), to assess the effectiveness of targeted cancer therapeutics in clear cell renal carcinoma cells via the interrogation of over 2880 drug–gene pairs (Giacosa et al. 2021), and to provide novel insights into circadian rhythm (Litovchenko et al. 2021) and evolution and development (Liu et al. 2020). Further increasing the scale of BRB-seq capabilities to optimize for higher throughput transcriptome analyses will only serve to increase the potential applications of this revolutionary technology.
References
-
Alpern, Daniel, Vincent Gardeux, Julie Russeil, Bastien Mangeat, Antonio C. A. Meireles-Filho, Romane Breysse, David Hacker, and Bart Deplancke. 2019. “BRB-Seq: Ultra-Affordable High-Throughput Transcriptomics Enabled by Bulk RNA Barcoding and Sequencing.” Genome Biology 20 (1): 71. https://doi.org/10.1186/s13059-019-1671-x.
-
Bast‐Habersbrunner, Andrea, Christoph Kiefer, Peter Weber, Tobias Fromme, Anna Schießl, Petra C. Schwalie, Bart Deplancke, Yongguo Li, and Martin Klingenspor. 2021. “LncRNA Ctcflos Orchestrates Transcription and Alternative Splicing in Thermogenic Adipogenesis.” EMBO Reports 22 (7): e51289.
-
Bevers, Roel P. J., Maria Litovchenko, Adamandia Kapopoulou, Virginie S. Braman, Matthew R. Robinson, Johan Auwerx, Brian Hollis, and Bart Deplancke. 2019. “Mitochondrial Haplotypes Affect Metabolic Phenotypes in the Drosophila Genetic Reference Panel.” Nature Metabolism 1 (12): 1226–42. https://doi.org/10.1038/s42255-019-0147-3.
-
Giacosa, Sofia, Catherine Pillet, Irinka Séraudie, Laurent Guyon, Yann Wallez, Caroline Roelants, Christophe Battail, et al. 2021. “Cooperative Blockade of CK2 and ATM Kinases Drives Apoptosis in VHL-Deficient Renal Carcinoma Cells through ROS Overproduction.” Cancers 13 (3): 576. https://doi.org/10.3390/cancers13030576.
-
Litovchenko, Maria, Antonio C. A. Meireles-Filho, Michael V. Frochaux, Roel P. J. Bevers, Alessio Prunotto, Ane Martin Anduaga, Brian Hollis, et al. 2021. “Extensive Tissue-Specific Expression Variation and Novel Regulators Underlying Circadian Behavior.” Science Advances 7 (5): eabc3781. https://doi.org/10.1126/sciadv.abc3781.
-
Liu, Jialin, Michael Frochaux, Vincent Gardeux, Bart Deplancke, and Marc Robinson-Rechavi. 2020. “Inter-Embryo Gene Expression Variability Recapitulates the Hourglass Pattern of Evo-Devo.” BMC Biology 18 (1): 129. https://doi.org/10.1186/s12915-020-00842-z.
-
Picelli, Simone, Åsa K. Björklund, Björn Reinius, Sven Sagasser, Gösta Winberg, and Rickard Sandberg. 2014. “Tn5 Transposase and Tagmentation Procedures for Massively Scaled Sequencing Projects.” Genome Research 24 (12): 2033–40. https://doi.org/10.1101/gr.177881.114.
-
Sena, Johnny A., Giulia Galotto, Nico P. Devitt, Melanie C. Connick, Jennifer L. Jacobi, Pooja E. Umale, Luis Vidali, and Callum J. Bell. 2018. “Unique Molecular Identifiers Reveal a Novel Sequencing Artefact with Implications for RNA-Seq Based Gene Expression Analysis.” Scientific Reports 8 (1): 13121. https://doi.org/10.1038/s41598-018-31064-7.
-
Ziegenhain, Christoph, Beate Vieth, Swati Parekh, Björn Reinius, Amy Guillaumet-Adkins, Martha Smets, Heinrich Leonhardt, Holger Heyn, Ines Hellmann, and Wolfgang Enard. 2017. “Comparative Analysis of Single-Cell RNA Sequencing Methods.” Molecular Cell 65 (4): 631-643.e4. https://doi.org/10.1016/j.molcel.2017.01.023.